Reaction of aldehydes or ketones to give β-hydroxy carbonyl compounds.
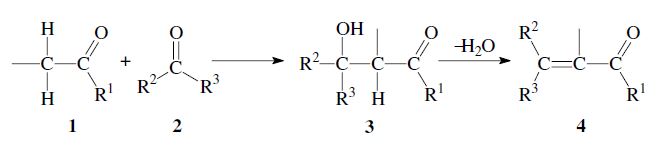
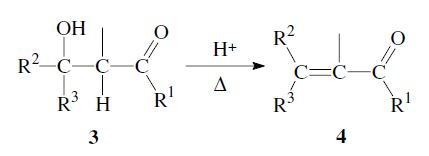
The addition of the α-carbon of an enolizable aldehyde or ketone 1 to the carbonyl group of a second aldehyde or ketone 2 is called the aldol reaction.1,2 It is a versatile method for the formation of carbon–carbon bonds, and is frequently used in organic chemistry. The initial reaction product is a β-hydroxy aldehyde (aldol) or β-hydroxy ketone (ketol) 3. A subsequent dehydration step can follow, to yield an α,β-unsaturated carbonyl compound 4. In that case the entire process is also called aldol condensation.
The aldol reaction as well as the dehydration are reversible. In order to obtain the desired product, the equilibrium might have to be shifted by appropriate reaction conditions (see below). The reaction can be performed with base catalysis as well as acid catalysis. The former is more common; here the enolizable carbonyl compound 1 is deprotonated at the α-carbon by base (e.g. alkali hydroxide) to give the enolate anion 5, which is stabilized by resonance:
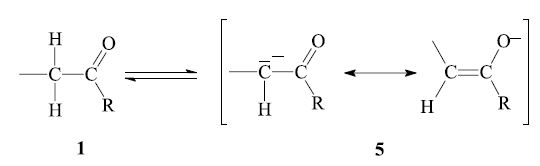
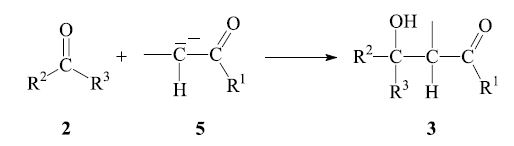
The next step is the nucleophilic addition of the enolate anion 5 to the carbonyl group of another, non-enolized, aldehyde molecule 2. The product which is obtained after workup is a β-hydroxy aldehyde or ketone 3:
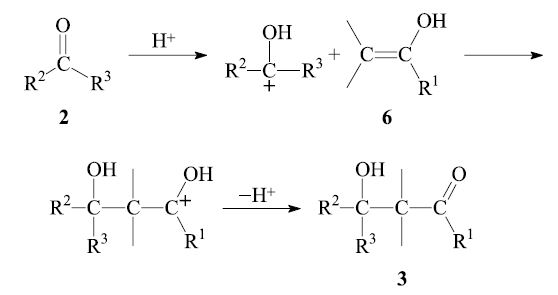
In the acid-catalyzed process, the enol 6 reacts with the protonated carbonyl group of another aldehyde molecule 2:
If the initially formed β-hydroxy carbonyl compound 3 still has an α-hydrogen, a subsequent elimination of water can take place, leading to an α,β-unsaturated aldehyde or ketone 4. In some cases the dehydration occurs already under the aldol reaction conditions; in general it can be carried out by heating in the presence of acid:
Several pairs of reactants are possible. The aldol reaction between two molecules of the same aldehyde is generally quite successful, since the equilibrium lies far to the right. For the analogous reaction of ketones, the equilibrium lies to the left, and the reaction conditions have to be adjusted properly in order to achieve satisfactory yields (e.g. by using a Soxhlet extractor).
With unsymmetrical ketones, having hydrogens at both α-carbons, a mixture of products can be formed. In general such ketones react preferentially at the less substituted side, to give the less sterically hindered product.
A different situation is found in the case of crossed aldol reactions, which are also called Claisen–Schmidt reactions. Here the problem arises, that generally a mixture of products might be obtained.
From a mixture of two different aldehydes, each with α-hydrogens, four different aldols can be formed—two aldols from reaction of molecules of the same aldehyde + two crossed aldol products; not even considering possible stereoisomers (see below). By taking into account the unsaturated carbonyl compounds which could be formed by dehydration from the aldols, eight different reaction products might be obtained, thus indicating that the aldol reaction may have preparative limitations.
If only one of the two aldehydes has an α-hydrogen, only two aldols can be formed; and numerous examples have been reported, where the crossed aldol reaction is the major pathway.2 For two different ketones, similar considerations do apply in addition to the unfavorable equilibrium mentioned above, which is why such reactions are seldom attempted.
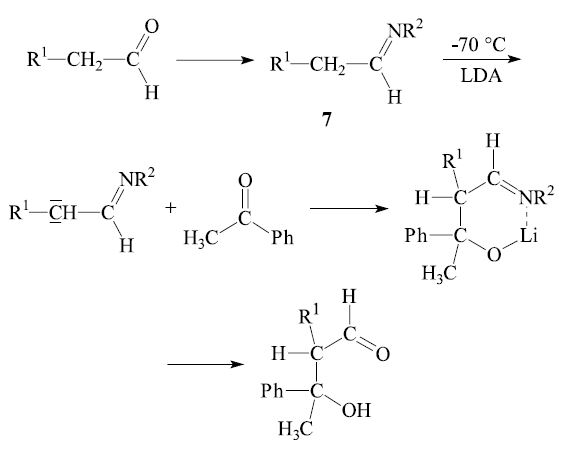
In general the reaction of an aldehyde with a ketone is synthetically useful. Even if both reactants can form an enol, the α-carbon of the ketone usually adds to the carbonyl group of the aldehyde. The opposite case—the addition of the α-carbon of an aldehyde to the carbonyl group of a ketone—can be achieved by the directed aldol reaction.3,4 The general procedure is to convert one reactant into a preformed enol derivative or a related species, prior to the intended aldol reaction. For instance, an aldehyde may be converted into an aldimine 7, which can be deprotonated by lithium diisopropylamide (LDA) and then add to the carbonyl group of a ketone:
By using the directed aldol reaction, unsymmetrical ketones can be made to react regioselectively. After conversion into an appropriate enol derivative (e.g. trimethylsilyl enol ether 8) the ketone reacts at the desired α-carbon.
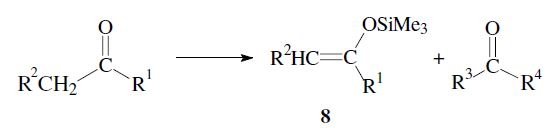
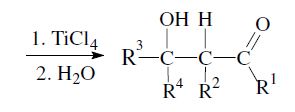
An important aspect is the control of the stereochemical outcome.5–7 During the course of the reaction two new chiral centers can be created and thus two diastereomeric pairs of enantiomers (syn/anti resp. erythro/threo pairs) may be obtained.
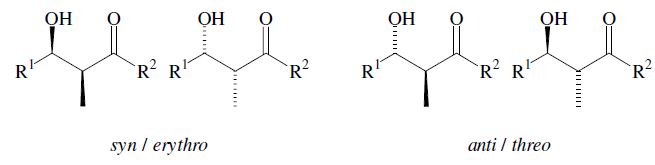
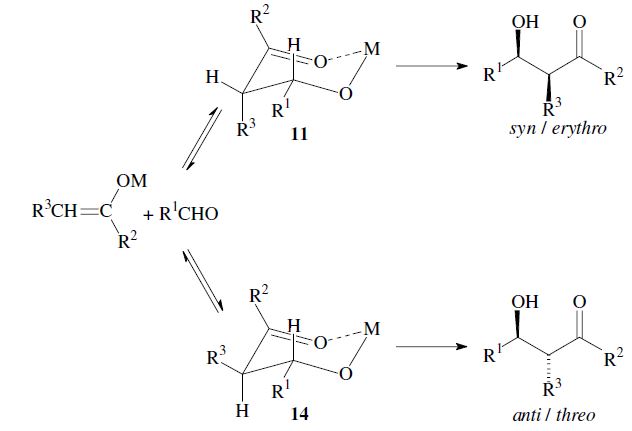
The enantiomers are obtained as a racemic mixture if no asymmetric induction becomes effective. The ratio of diastereomers depends on structural features of the reactants as well as the reaction conditions as outlined in the following. By using properly substituted preformed enolates, the diastereoselectivity of the aldol reaction can be controlled.7 Such enolates can show E-or Z-configuration at the carbon–carbon double bond. With Z-enolates 9, the syn products are formed preferentially, while E-enolates 12 lead mainly to anti products. This stereochemical outcome can be rationalized to arise from the more favored transition state 10 and 13 respectively:
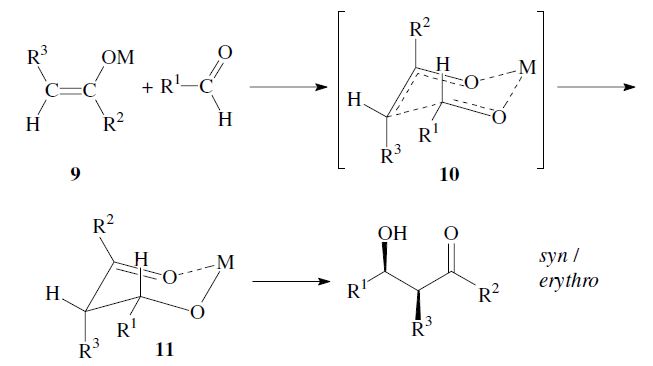
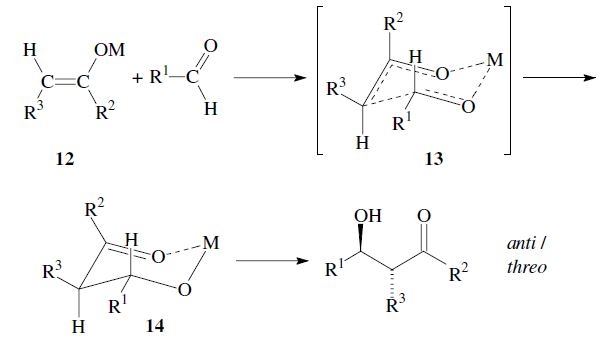
Under conditions which allow for equilibration (thermodynamic control) however, the anti -product is obtained, since the metal-chelate 14 is the more stable. As compared to 11 it has more substituents in the favorable equatorial position:
With an appropriate chiral reactant, high enantioselectivity can be achieved, as a result of asymmetric induction.8 If both reactants are chiral, this procedure is called the double asymmetric reaction,6 and the observed enantioselectivity can be even higher.
An enantioselective aldol reaction may also be achieved with non-chiral starting materials by employing an asymmetric Lewis acid as catalyst:9
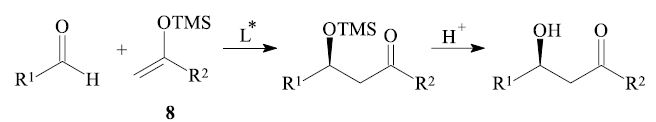
For example in the so-called Mukaiyama aldol reaction4,10,11 of an aldehyde R1-CHO and a trimethylsilyl enol ether 8, which is catalyzed by Lewis acids, the required asymmetric environment in the carbon–carbon bond forming step can be created by employing an asymmetric Lewis acid L* in catalytic amounts.
Especially with the ordinary aldol reaction a number of side reactions can be observed, as a result of the high reactivity of starting materials and products. For instance, the α,β-unsaturated carbonyl compounds 4 can undergo further aldol reactions by reacting as vinylogous components. In addition compounds 4 are potential substrates for the Michael reaction.
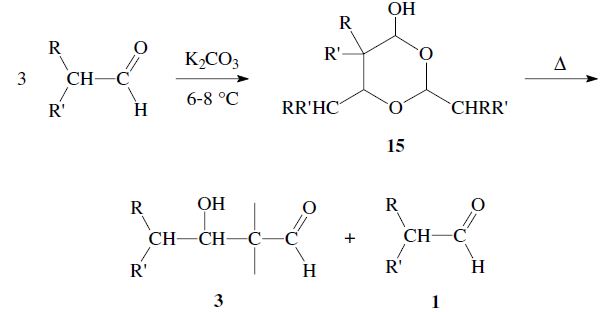
Aldehydes can react through a hydride transfer as in the Cannizzaro reaction. Moreover aldoxanes 15 may be formed; although these decompose upon heating to give an aldol 3 and aldehyde 1:
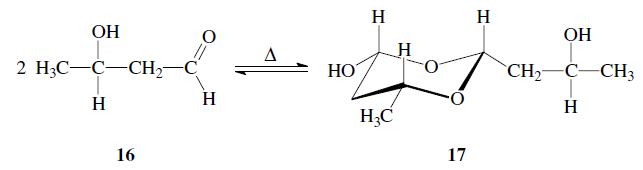
Aldols can form dimers; e.g. acetaldol 16 dimerizes to give paraldol 17:
Because of the many possible reactions of aldols, it is generally recommended to use a freshly distilled product for further synthetic steps. Besides the aldol reaction in the true sense, there are several other analogous reactions, where some enolate species adds to a carbonyl compound. Such reactions are often called aldol-type reactions; the term aldol reaction is reserved for the reaction of aldehydes and ketones.
References
- M. A. Wurtz, Bull. Soc. Chim. Fr. 1872, 17, 436–442.
- A. T. Nielsen, W. J. Houlihan, Org. React. 1968, 16, 1–438.
- G. Wittig, H. Reiff, Angew. Chem. 1968, 80, 8–15; Angew. Chem. Int. Ed. Engl. 1968, 7, 7.
- T. Mukaiyama, Org. React. 1982, 28, 203–331;
T. Mukaiyama, S. Kobayashi, Org. React. 1994, 46, 1–103. - C. H. Heathcock, Science 1981, 214, 395–400.
- S. Masamune, W. Choy, J. S. Petersen, L. S. Sita, Angew. Chem. 1985, 97, 1–31; Angew. Chem. Int. Ed. Engl. 1985, 24, 1.
- C. H. Heathcock in Modern Synthetic Methods 1992 (Ed.: R. Scheffold), VHCA, Basel, 1992, p. 1–102.
- D. Enders, R. W. Hoffmann, Chem. Unserer Zeit 1985, 19, 177–190.
- U. Koert, Nachr. Chem. Techn. Lab. 1995, 43, 1068–1074.
- S. Kobayashi, H. Uchiro, I. Shiina, T. Mukaiyama, Tetrahedron 1993, 49, 1761–1772.
- T. D. Machajewski, C. H. Wong, Angew. Chem. 2000, 112, 1406–1430; Angew. Chem. Int. Ed. Engl. 2000, 39, 1376.